Introduction
Neutropenia and elevated concentrations of hepatic transaminases are commonly found among children visiting emergency departments (EDs) or outpatient departments (OPDs). When these symptoms manifest in previously healthy children, the symptoms are often presumed to stem from viral infections (1,2). If the symptoms persist, consulting a metabolism specialist offers the children an opportunity for early diagnosis of an inborn error of metabolism, potentially preventing its complications.
Glycogen storage disease type (GSD) Ib is a rare inborn error of glucose metabolism resulting from biallelic mutations in the SLC37A4 gene, encoding glucose-6-phosphate transporter (3). Dysfunction of the transporter may lead to the accumulation of 1,5-anhydroglucitol-6-phosphate (1,5-AG6P) within the granulocytes, inducing neutrophil dysfunction and impairing glucose metabolism, ultimately resulting in apoptosis of neutrophils (4). As a result, morbid children can develop symptoms such as neutropenia, elevated hepatic transaminases, hepatomegaly, hypoglycemia, or growth retardation. The neutropenia can cause recurrent infections, impaired wound healing, and mucosal ulceration (5).
The standard treatment for GSD Ib-associated neutropenia was granulocyte-colony stimulating factor (G-CSF). This therapeutic approach has been changed since a clinical trial conducted in 2020 on the off-label use of empagliflozin, a sodium-glucose co-transporter-2 inhibitor, demonstrated the improvement of neutropenia and neutrophil dysfunction in GSD Ib by reducing 1,5-AG6P entry into the neutrophils (6). In detail, among the 89 patients (median age of 10.5 years [range, 0-38]) who had been on G-CSF before starting empagliflozin therapy, 49 (55.1%) stopped and 15 (16.9%) received a reduced dose of G-CSF, respectively, with 20 (17.9%) undergoing adverse effects (AEs) of empagliflozin such as hypoglycemia (6). Here, we report 2 cases of Korean adolescents with GSD Ib-associated neutropenia successfully treated with empagliflozin. This study was exempt from review by the institutional review board of Kyungpook National University Chilgok Hospital with a waiver for informed consent (IRB no. KNUCH 2024-03-035).
Case
1. Case 1
A previously healthy 17-year-old boy presented to the ED with abdominal discomfort, frequent epistaxis, easy bruising, and chronic myalgia. At the age of 9 years, he underwent abdominal distension accompanied by leukopenia and normal concentrations of hepatic transaminases. However, further workup was not pursued at that time, and no hypoglycemic episodes were reported. At the ED, his laboratory findings were as follows: white blood cells (WBCs), 2,500/╬╝L; absolute neutrophil count (ANC), 550/╬╝L; alanine aminotransferase (ALT), 50 U/L; lactic acid, 4.2 mmol/L (reference value, < 2); uric acid, 10.4 mg/dL (3.1-7.9); total cholesterol, 274 mg/dL (< 200); and triglyceride, 876 mg/dL (< 150). Liver magnetic resonance imaging showed multiple small hepatocellular adenomas, and liver biopsy indicated steatosis without malignancy, consistent with GSD Ib. Genetic testing identified compound heterozygous pathogenic mutations of c.148G>A (p.Gly50Arg); c.443C>T (p.Ala148Val) in the SLC37A4 gene, confirming a diagnosis of GSD Ib.
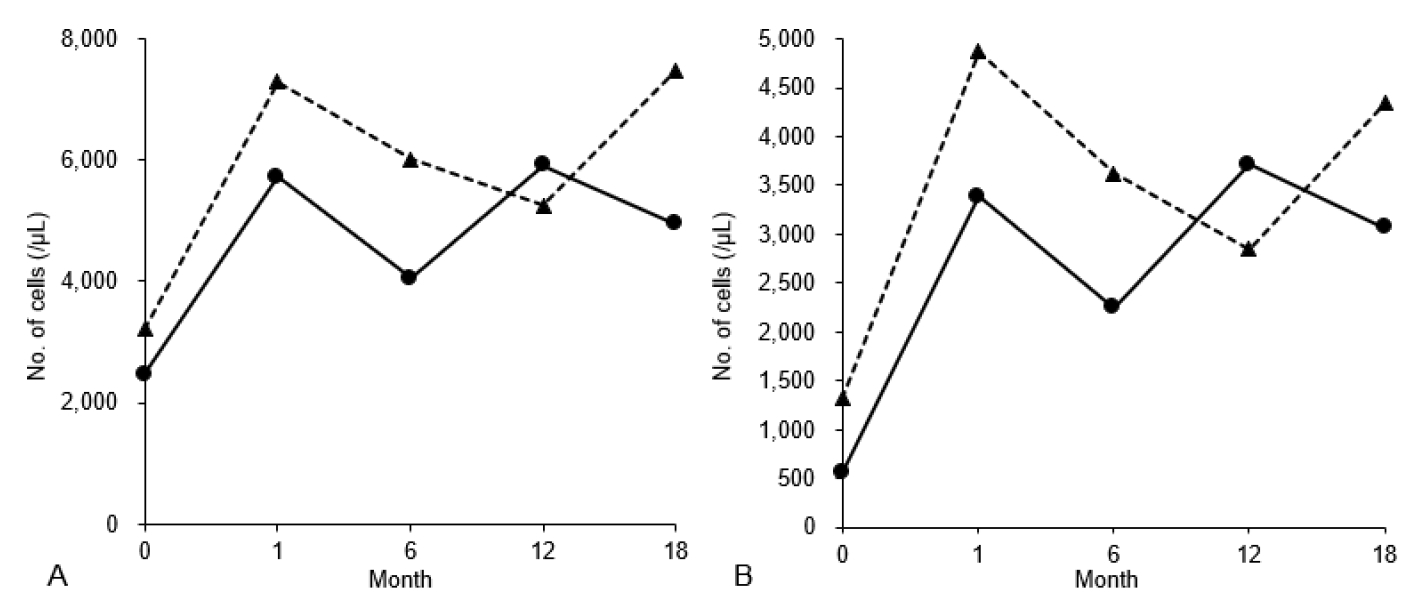
The boy adhered to a diet with uncooked corn starch and restricted refined sugar. A computed tomography scan showed a small, asymptomatic nephrolithiasis in the right kidney. He was also administered atorvastatin for his dyslipidemia, allopurinol for hyperuricemia, and potassium citrate for nephrolithiasis. Instead of administering G-CSF, empagliflozin was initiated with 1 dose of 5 mg daily, gradually increased to 2 doses of 10 mg daily (0.29 mg/kg/day divided into 2 doses) over 4 days. The values of WBCs and ANC have been normalized over the period of 18 months (Fig. 1). Within 1 month of starting the medication, he reported improvement in the self-reported symptoms. No infection was reported during the follow-up period. Glycosuria was detected during empagliflozin therapy, with the renal function remained intact.
2. Case 2
A 15-year-old girl presented with proteinuria and elevated ALT to the OPD. She was diagnosed with GSD Ib at the age of 15 months after an accidental detection of the laboratory abnormalities during a workup for community-acquired pneumonia. At the time of diagnosis, the genetic test had proved a pathogenic and likely pathogenic compound heterozygous mutations in the SLC37A4 gene: c.443C>T (p.Ala148Val) and c.618G>A (p.Gly273Asp). At the ED, she was prescribed fenofibrate and allopurinol and followed a conventional dietary management with uncooked corn starch. Blood and urine findings were as follows: WBCs, 3,190/╬╝L; ANC, 1,230/╬╝L; hemoglobin, 11.7 g/dL; ALT, 67 U/L; lactic acid, 3.5 mmol/L; uric acid, 7.0 mg/dL; total cholesterol, 226 mg/dL; triglyceride, 394 mg/dL; blood urea nitrogen, 9.9 mg/dL; creatinine, 0.4 mg/dL; and urine protein/creatinine ratio, 1.3.
After hospitalization, a renal biopsy proved a focal collection of glycogen particles in the proximal tubular epithelial cells without any findings of glomerular injury, indicating GSD Ib-associated proteinuria. Losartan was prescribed with continued monitoring of renal function. Due to the persistent leukopenia, empagliflozin was administered at a dose of 5 mg once daily, and the dose was increased to 15 mg/day over 3 days (0.26 mg/kg/day divided into 2 doses; 10 mg in the morning and 5 mg in the evening). After 1 month, values of WBCs and ANC have been normalized (Fig. 1). After using empagliflozin, glycosuria was detected on a urine dipstick test. Throughout the 18-month observation period, no infection was reported while the girl consistently adhered to a treatment regimen of fenofibrate and empagliflozin.
Discussion
To our knowledge, this article presents the first documented cases of empagliflozin therapy for GSD Ib in Korea. Our observations align with global research, demonstrating a favorable increase in ANC over an 18-month period with no reported AEs (7-9). Despite the unclear long-term outcomes, early detection in EDs, which can be facilitated by expert referral in cases of neutropenia with elevated hepatic transaminases, may improve the outcomes.
After presenting to the ED with neutropenia, patient 1 was referred to a specialist, which led us to implement a novel therapeutic approach. After the therapy, there was improvement in the abovementioned symptoms as well as in ANC, suggesting a positive impact on the boyέΑβs quality of life. The symptoms may be related to neutrophil dysfunction or early manifestations of GSD Ib-related inflammatory bowel disease, potentially linked to therapeutic efficacy in restoring neutrophil functions (7,10). Metabolic derangements, such as dyslipidemia and hyperuricemia, also contribute to the disease burden. Patient 1 was treated with allopurinol, while patient 2 showed a normal concentration of uric acid, eliminating the need for allopurinol after empagliflozin therapy. Further research in this aspect is needed as patient cohorts and global experience expand.
GSD Ib may progress while showing various clinical manifestations. None of our patients were administered G-CSF due to concerns about its AEs, ranging from mild bone pain to an increased risk of myelodysplastic syndrome or hematologic malignancy (11,12).
Since the initial report in 2020 by Wortmann et al. (13), the effectiveness of empagliflozin in treating GSD Ib has been documented in subsequent case reports, consistently highlighting its benefits in improving neutropenia, neutrophil function, and overall outcomes (7). In our cases, medication adjustments were made in the OPD. The initial dose for all cases was 5 mg once daily, gradually increasing by 5 mg/day to reach the target dose. In case 1, the dosage was increased to 10 mg twice daily, and in case 2, 15 mg/day. Considering the mechanism of action, a higher daytime dose is recommended due to the potential increased urination and thirst. Our dosage was slightly lower than a previously reported median pediatric dose (0.4 mg/kg/day divided into 2 doses) (6). Both patients tolerated the medication, with no reported AEs, such as hypoglycemia, dehydration, or renal insufficiency.
Founder effects related to genotype differences in the SLC37A4 gene have been well-studied among various races. Similar genotypes are frequently observed in Korea, China, and Japan, compared to Caucasian, Hispanic, and Jewish populations. The identified variants of the gene in the present cases are all classified as pathogenic or likely pathogenic according to the classification of the American College of Medical Genetics and Genomics and are previously reported mutations. Variants of c.148G>A (p.Gly50Arg), c.443C>T (p.Ala148Val), and c.618G>A (p.Gly273Asp) were reported in Korean GSD Ib patients (14-17). From a genotype-phenotype matching perspective, instances of neutropenia were found in patients with all 3 variants although not all GSD Ib cases exhibit neutropenia (9,18).
Glycosuria is induced by empagliflozin therapy due to its mechanism of action (19). Despite the safety profiles studied in clinical trials for type 2 diabetes mellitus, long-term outcomes required evaluation for sustained treatment (19). Therefore, it is essential to closely monitor the renal function and symptoms suggestive of gastroenteritis or dehydration in patients with GSD Ib. With the monitoring, empagliflozin would be administered continuously unless our patients present with gastroenteritis or febrile infections, or are at risk of dehydration. Previous reports emphasized the effective management of urinary tract infections in women with empagliflozin-induced glycosuria. A limitation of our study was the lack of measurement of 1,5-AG6P, a metabolite indicating the progression of GSD Ib. For practical reasons, we instead assessed glycosuria using conventional urine dipsticks.
As children with GSD Ib are in transition into adolescence or adulthood, they remain at risk of severe, chronic complications such as renal dysfunction, osteoporosis, cardiovascular disease, inflammatory bowel disease, or hepatocellular malignancy (5). This emphasizes the importance of lifelong monitoring and multidisciplinary management to optimize the outcomes. Further research in children under 10 years is needed to investigate the feasibility of empagliflozin as a preventive first-line therapy. Comprehensive studies are required to evaluate the long-term benefits and safety of empagliflozin in managing GSD Ib, facilitating its integration into clinical practice.
Diagnosing GSD Ib is complex and usually initiated by a referral from EDs or OPDs when neutropenia or elevated hepatic transaminases are detected. The novel empagliflozin therapy demonstrated the restoration of counts and function of the neutrophils. It appears to be a safe and effective therapeutic option for children with GSD Ib and associated neutropenia.